
Všeobecné
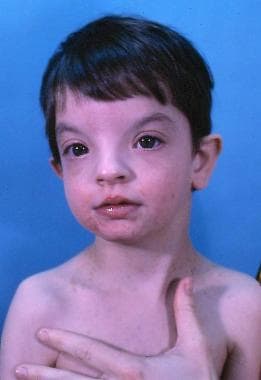
Wolf-Hirschhornov syndróm (WHS) je extrémne zriedkavá chromozomálna porucha spôsobená chýbajúcim kúskom (čiastočná delécia alebo monozómia) krátkeho ramienka chromozómu 4. Medzi hlavné príznaky môžu patriť naširoko "posadené" oči (očný hypertelorizmus) so širokým alebo zobákovitým nosom, malá hlava (mikrocefália), zle posadené uši, spomalený rast, srdcové defekty, problémy s intelektom. Príznaky tohto syndrómu sa líšia od človeka k človeku na základe veľkosti a umiestnenia chýbajúcej časti chromozómu 4.
Diagnostika:
Chromozómy sa nachádzajú v jadre všetkých buniek tela. Nesú genetické vlastnosti každého jednotlivca. Páry ľudských chromozómov sú očíslované od 1 do 22, pričom 23. pár je tvorený z chromozómov X a Y, pričom XX je spojené so ženským pohlavím a XY spojené s mužským pohlavím. Každý chromozóm má krátke rameno označené „p“ a dlhé rameno označené ako „q“. Chromozómy sa ďalej delia na očíslované pásma. Napríklad „chromozóm 4p16.3“ (vyslovuje sa „štyri p jeden až šesť bodov tri“) označuje pásmo 16.3 na krátkom ramene chromozómu 4. Číslované pásy sa používajú na určenie polohy génov nachádzajúcich sa na každom chromozóme.
Túto poruchu spôsobuje delécia časti krátkeho ramena chromozómu 4 (4p). Verí sa, že časť pásma 16.3 na chromozóme 4p (4p16.3) je „kritickou oblasťou“ pre poruchu, čo znamená, že delécia tejto oblasti vedie k úplnej expresii Wolf-Hirschhornovho syndrómu. U väčšiny ľudí sa delécia spôsobujúca WHS vyskytla extrémne skoro vo vývoji tohto jedinca (pravdepodobne vo vajíčku alebo spermiách pred oplodnením) a nezdedí sa od rodičov tejto osoby. Oveľa menej často sa porucha dedí po rodičovi, ktorý má vyváženú translokáciu. WHS je extrémne zriedkavá porucha. Štúdie vykonané pred asi 25 rokmi naznačili, že porucha sa vyskytla u približne 1 z približne 50 000 živo narodených detí s pomerom žien k mužom 2: 1. Novšie štúdie naznačujú, že frekvencia poruchy je podhodnotená z dôvodu nesprávnej diagnózy.
Diagnózu WHS môže naznačovať charakteristický vzhľad tváre, zlyhanie rastu či oneskorenie vývoja. Diagnóza je potvrdená detekciou delécie kritickej oblasti Wolf-Hirschhornovho syndrómu (WHSCR) cytogenetickou (chromozómovou) analýzou. Konvenčná cytogenetická analýza detekuje menej ako polovicu delécií, ktoré spôsobujú WHS. Fluorescenčná in situ hybridizácia (FISH) s použitím sondy WHSCR má oveľa lepšiu mieru detekcie ako štandardný karyotyp a bude detekovať väčšinu pacientov. Diagnostickým testom podľa voľby je však chromozomálny mikročip, ktorý detekuje v podstate všetky delécie WHSCR a definuje veľkosť delécie. Chromozomálna mikromatica môže nájsť aj ďalšie prešmyky chromozómov, napríklad ďalšie kúsky ďalších chromozómov, ktoré sa vyskytujú u mnohých pacientov s WHS.
Liečba:
Pretože WHS je veľmi variabilný syndróm, liečba a intervencie musia byť šité na mieru potrebám postihnutého jedinca. Pacienti so známou alebo podozrivou diagnózou WHS by mali mať komplexné hodnotenie od skúseného genetika. Väčšina pacientov bude musieť navštíviť niekoľko špecialistov. Pacienti by mali čo najskôr po diagnostikovaní podstúpiť neurologické vyšetrenie, podrobné kardiologické (srdcové) vyšetrenie, očné vyšetrenie, vyšetrenie sluchu, obličiek, vyhodnotenie výživy a vývojové vyšetrenie. Funkcia obličiek musí byť neustále sledovaná. Všetci jedinci potrebujú komplexnú vývojovú a rehabilitačnú podporu vrátane: kŕmenia, asistenčnej komunikácie, reči, fyzikálnej terapie, pracovnej terapie a školskej podpory. Genetické poradenstvo sa odporúča rodinám detí s Wolf-Hirschhornovým syndrómom.
- spomalený rast
- hypotónia
- malá hlava (mikrocefália), "prepadnuté" oči
- abnormality pohlavných orgánov
- problémy s kosťami a zubami
- problémy s imunitou
- problémy s kŕmením
- možnosť problémov s intelektom (ľahké až ťažké formy)
Pre spojenie sa s ďalšími ľuďmi s rovnakou diagnózou vo vašom okolí sa prihláste.
Prihlásenie