Urbachov-Wietheho choroba
Find a cause you want to help. Every contribution counts.

Všeobecné
Urbachov-Wietheho syndróm (anglicky Urbach-Wiethe syndrome / lipoidy proteinosis / hyalinosis cutis et mucosae) je zriedkavé, autozomálne recesívne geneticky podmienené ochorenie postihujúce kožu a nervovú sústavu. Dôsledkom mutácie na chromozóme 1 dochádza k strate funkcie ECM1 génu a k tvorbe hyalínu podobné stavebné látky v koži. Následne koža hrubne a tvrdne. V mozgu sú často nájdené kalcifikácie spôsobujúce rôzne neurologické prejavy choroby.
Diagnostika:
Urbachov-Wietheho syndróm je typicky diagnostikovaný na základe pre neho charakteristických prejavov na koži. Možné je testovať aj samotnú kožu na prítomnosť hyalínu podobné látky. CT snímka nám ozrejmí prítomnosť kalcifikácie v mozgu, ktoré však nemusia s týmto stavom priamo súvisieť. Jednoznačné potvrdenie diagnózy je možné vďaka genetickému testovaniu na mutáciu ECM1 génu.
Liečba:
V súčasnej dobe je tento syndróm neliečiteľný (rovnako ako iné genetické ochorenia), ale konkrétne prejavy možno relatívne dobre kompenzovať. Nejedná sa o život ohrozujúce ochorenia a pri rýchlom riešení respiračných aj iných komplikácií ani neskracuje život pacienta.
- porcelánový vzhľad kože
- ľahko poraniteľná koža
- pľuzgiere a jazvy po drobných poraneniach
- suchá a zvráskavená koža
- biele alebo žlté infiltráty na perách, sliznici dutiny ústnej, mandliach ...
- infekcia horných dýchacích ciest, niekedy je nutné zaviesť tracheostómiu na zmiernenie symptómov
- možný problém reči
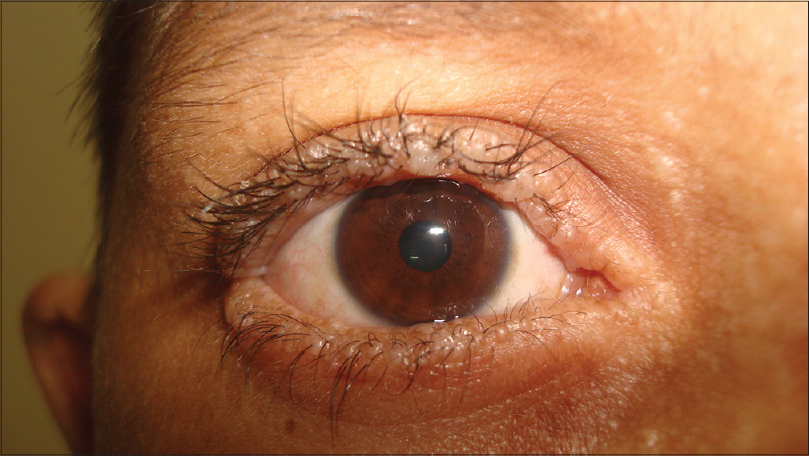
- pupence (výrastky) na okraji očných viečok sú typické
- vypadávanie vlasov
- rohovkový vred
- fokálna degenerácia makuly a teda porucha vízu
- zápal ďasien
- poruchy vytvárania a ukladania spomienok spojených s emocionálnymi udalosťami
- neschopnosť rozpoznávania reakcií na podnety spojené hlavne s nepríjemnou udalosťou
- absencia reakcie strachu ako je stŕpnutie (nehybnosť), tachykardia (rýchly srdcový tep), zvýšené dýchanie, zovretie žalúdka a vylučovanie stresových hormónov
- epilepsia
- ataky úzkosti
- poruchy nálady
- psychotické poruchy a ďalšie možné psychiatrické prejavy
- chrapľavý hlas
- slabý plač novorodenca
Pre spojenie sa s ďalšími ľuďmi s rovnakou diagnózou vo vašom okolí sa prihláste.
Prihlásenie