
Všeobecné
Crouzonov syndróm, predtým tiež označovaný ako kraniofaciálna dyzostóza (dysostosis craniofacialis) je vrodená porucha vývoja lebky s autozomálne dominantnou dedičnosťou. Boli popísané asociácie s mutáciami v génoch FGFR2 a FGFR3, ktoré kódujú receptory pre rastové faktory fibroblastov.
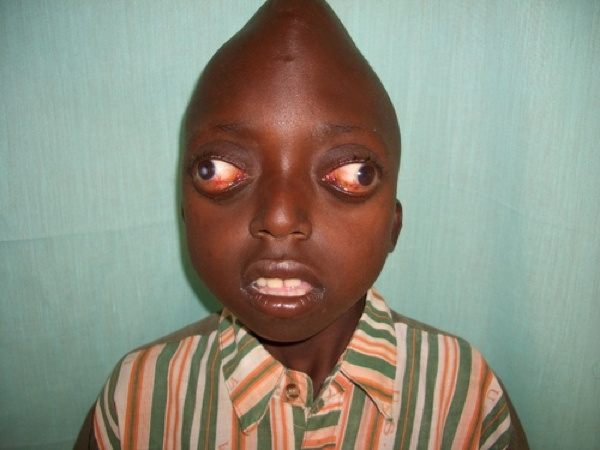
Hlavným príznakom je vežovitá lebka (turicefalia) so širokým čelom, prípadne s vysokým hrboľom v oblasti veľkej fontanely. Dôvodom je predčasný zrast šípového švu. Ďalšími príznakmi sú: exoftalmus (vystúpené očné bulby), orbitálny hypertelorismus (široko od seba postavené oči), ľahko antimongoloidné postavenie očných štrbín (os očných viečok smeruje od stredu smerom nadol), divergentný strabizmus (rozbiehavé škúlenie), hypoplázia maxilou (menšie horná čeľusť ), tzv. "nos papagájového zobáka".
Porucha rastu lebky môže viesť k útlaku optického nervu a jeho následnej atrofie. Z rovnakých dôvodov môže dôjsť aj k poruchám sluchu.
Liečba
Crouzonov syndróm je možné riešiť operáciou lebky v ranom detstve. Ak je operácia vykonaná, jedinci sa dožívajú normálneho veku. Aj po operácii je bežný podkus (opak predkusu), tieto osoby majú problém so žuvaním potravy a radšej si ju krájajú alebo lámu.
- vežovitá lebka
- široké čelo (poprípade vysoký hrboľ v oblasti veľkej fontanely)
- exoftalmus (vystúpené očné bulby)
- orbitálny hypertelorismus (široko od seba postavené oči)
- ľahko antimongoloidné postavenie očných štrbín (os očných viečok smeruje od stredu smerom nadol)
- divergentný strabizmus (rozbiehavé škúlenie)
- hypoplázia maxilou (menšia horná čeľusť)
Pre spojenie sa s ďalšími ľuďmi s rovnakou diagnózou vo vašom okolí sa prihláste.
Prihlásenie