
Všeobecné
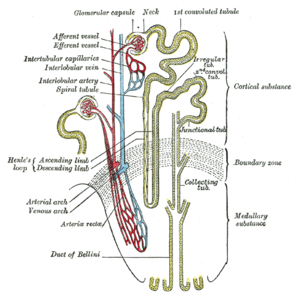
Bartterov syndróm (angl. Bartter syndrome) je vrodená tubulopatia, čiže ochorenie obličiek – konkrétne jeho časti nefrónu v oblasti vzostupného ramienka na Henleho kľučke, pri ktorom je narušená ich resorpčná schopnosť (porucha spätného vstrebávania minerálov v obličkách), čo spôsobuje kombináciu poruchy vodného a elektrolytového metabolizmu. Syndróm vzniká ako dôsledok poruchy tubulárneho transportu a vylučovania iónov. Je spôsobená mutáciou niekoľkých génov pre niekoľko transportných systémov v Henleho kľučke - sodíkovo - draslíko-chloridového kotransportéru (Na-K-2Cl), draslíkového a chloridového kanála. Dôsledkom sú nadmerné straty minerálov v moči (sodíka, draslíka a chloridov) následkom toho vzniknutá hypokaliémia, hypochloremická metabolická alkalóza v krvi - (tj. porucha vnútorného prostredia). Incidencia tohto ochorenia (výskyt v populácii v Európe) je 1/1000 000. Znížená hodnota sodíka v macula densa (aktívna časť nefrónu, ktorá reguluje krvný tlak pomocou zvyšovania alebo znižovania hormónov) zvýši aktivitu RAAS (spleť hormónov - renín, angiotenzín, aldosterón), následkom toho sa zvyšuje hormón aldosterón a dochádza tak k dg. sekundárnemu hyperaldosteronizmu so všetkými klinickými prejavmi (krvný tlak je však v tomto prípade normálny). U niektorých pacientov sa pridružuje mentálna retardácia alebo postupné zlyhávanie obličiek.
Príčiny:
Syndróm je dôsledkom genetickej mutácie niekoľkých génov pre niekoľko transportných systémov minerálových kanálov. Toto ochorenie je vrodené. Existuje viac typov porúch, ktoré potom spôsobujú rôzne ťažké formy choroby. Choroba sa zvyčajne začne prejavovať v ranom alebo neskorom detstve. Dedičnosť je autozomálne recesívna, čiže na prejav ochorenia sú potrebné 2 postihnuté gény od oboch rodičov - dedičnosť je v tomto prípade raritná.
Príznaky a diagnostika:
Podozrenie možno získať z nižšie uvedených príznakov, v krvných vzorkách nájdeme zníženú koncentráciu základných iónov, najmä draslíka, prítomná je aj porucha vnútorného prostredia - metabolická alkalóza v krvi. V moči nájdeme zvýšený odpad minerálov. Pri vyšetrení acidobázickej rovnováhy v krvi nachádzame alkalózu. Chorobu a jej konkrétny typ potom môže odhaliť špeciálne genetické vyšetrenie.
Poznáme 6 podtypov Barrterovho syndrómu podľa poškodenia kanálov
- Typ I − defekt Na+/K+/2Cl--kotransportéru (NKCC2, gén SLC12A); manifestuje sa už v kojeneckom veku, väčšinou sú to deti, ktoré sú predčasne narodené, v tehotenstve bol diagnostikovaný polyhydramniom (nadmerné množstvo plodovej vody)
- Typ II − defekt ATP-závislého apikálnehodraslíkového kanálu (ROMK1, gen KCNJ1); fenotypicky je rovnaký ako typ I.
- Typ III − defekt bazolaterálneho chloridového kanálu (ClC-Kb, gen CLCNKB); u 30 % pacientov je prítomná hypomagnezémia (u typu I a II neprítomné).
- Typ IVa − defekt β-podjednotky bazolaterálneho chloridového kanálu (Barttin, gén BSND); charakteristická triáda: Bartterov syndrom, obličkové zlyhávanie, porucha sluchu.
- Typ IVb − kombinovaná dysfunkcia dvoch chloridových kanálov ClC-Ka a ClC-Kb (gény CLCNKA a CLCNKB), prenatálna manifestácia, polyhydramnion
- Typ V − transientná forma (defekt v géne MAGED2), polyhydramnion v tehotenstve, excesívna strata solí so sekundárnou metabolickou alkalózou, spontánne vymizne v prvých mesiacoch života
Diagnostika:
Na základe klinických príznakov, poruchy elektrolytov a vnútorného prostredia rozbehne lekár širokú diferenciálnu diagnostiku za účelom dodiagnostikovania tubulopatie a poruchy elektrolytov (najčastejšie sa to deje počas hospitalizácie na Internej klinike - NÚDCH), napomôcť môžu laboratórne výsledky krvi, moču, ultrazvukového vyšetrenia, avšak definitívnu diagnózu stanoví genetické vyšetrenie.
Liečba:
Vyliečiť chorobu nevieme, liečba je symptomatická. Dôležité je pacientom dodávať dostatok tekutín a minerálov, ktoré vyrovnajú ich straty močom. Dôležité sú teda pravidelné kontroly mineralogramu v krvi cestou obvodného lekára. V niektorých prípadoch môže s postupom času dôjsť k zlyhávaniu obličiek - preto je pacient kontrolovaný nefrológom, v terminálnom zlyhaní obličiek je pacient zaradený do dialyzačného programu.
- postihnutie obličkových kanálikov, ktoré slúžia k spätnému vstrebávaniu iónov a tekutín z prvovytvoreného moču. To vedie k nadmerným stratám niektorých minerálov do moču (napr. draslíka, sodíka, vápnika a chlóru) a tiež k stratám vody. V niektorých prípadoch dochádza k zlyhávaniu obličiek.
- zvýšené hladiny RAAS a aldosterónu a vzniká sekundárny hyperaldosteronizmus aj s prítomnými klinickými prejavmi (krvný tlak je normálny)
- nadmerné močenie s veľkými stratami tekutín a s tým súvisiacím smädom. Nedostatok tekutín vedie k dehydratácii, prítomná býva únava, znížená výkonnosť a sklon k nízkemu tlaku. Straty draslíka vedú zvyčajne k zníženej koncentrácii draslíka v krvi (hypokalémia) so všetkými jej dôsledkami (svalová slabosť, poruchy srdcového rytmu a pod).
- niektoré formy choroby sú spojené aj so zvýšenými stratami vápnika do moču (hyperkalciúria). Vápnik potom môže začať vytvárať močové kamene. Nedostatok vápnika v krvi potom môže spôsobiť svalové kŕče.
- poruchy rastu
- metabolická alkalóza v krvi
- možná mentálna retardácia
Pre spojenie sa s ďalšími ľuďmi s rovnakou diagnózou vo vašom okolí sa prihláste.
Prihlásenie