
Všeobecné
Alagillov syndróm (angl. Allagile syndrome) je veľmi zriedkavé geneticky podmienené vrodené ochorenie. Vyskytuje sa približne u 1 zo 70 000 - 100 000 živo narodených detí. Poškodzuje viaceré orgány - pečeň, srdce, obličky, skelet, oči. Forma tohto syndrómu je veľmi variabilná, závažnosť je v širokej škále.
Príčiny:
Príčinou je genetická mutácia JAG1 génu (zodpovedná za 88% prípadov) alebo NOTCH2 génu. Mutácia vyniká počas vývoja daného jedinca de novo, alebo je zdedená od rodičov dominantne autozomálne. Mutácia poškodzuje gén, ktorý je zodpovedný za správnu tvorbu vnútornej štruktúry tela počas vývoja plodu. Z tohto dôvodu sú dôsledkom tejto poruchy početné deformácie a chyby tkanív aj orgánov. Vznik mutácie alebo dedičnosť je rovnaká ako u muža, tak aj u žien.
Prejavy:
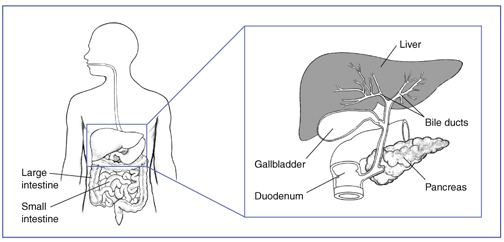
Už u malých detí počas prvých štyroch mesiacov možno diagnostikovať žltačku, svrbenie kože (pruritus), rôzne štádium poškodenia pečene, cholestázu (z dôvodu nedostatku žlčových kanálikov a nemožnosť odtoku žlče). Asi u 90% pacientov s Alagillovým syndrómom je diagnostikované chýbanie žlčových kanálikov na odtok žlče do čreva. Stolica býva svetlá, moč má tmavú farbu. Novorodenci trpia poruchami trávenia - najmä tukov, porucha vstrebávania vitamínov (A,D,E,K) a mikronutrientov a z toho vyplyvajúce príznaky (porucha kostí, porucha zraku, etc). Novorodenci neprospievajú, trpia malabsorbpciou, prípadne rozvratom vnútorného prostredia. Môže byť prítomný ľahký oneskorený psychomotorický vývoj. Poškodenie pečene môže progredovať až do cirhózy a zlyhania pečene (asi 15% pacientov s touto dg). Pankreas má tiež nedostatočnú funkciu (enzymatickú ako aj hormonálnu).
Už u novorodencov môžeme klinicky zachytiť šelest na srdci. Srdcové poškodenia bývajú v rozsiahlej škále od benígneho (nevýznameho) šelestu na srdci po závažné a život ohrozujúce defekty srdca, ktoré treba riešiť chirurgicky. Prítomné bývajú defekty komorového septa, zúženie pľúcnych tepiev v rôznom rozsahu, fallotova tetralógia, porucha rytmu - WPW syndróm. Poškodenie srdca môže spôsobovať omodrenie pier, tváre pri zvýšenej potrebe kyslíka, pri námahe, pri ťažších poruchách aj v pokoji.
Typickým príznakom pre Alagillov syndróm býva posteriórny embryotoxón, čo je zhrubnutý prstenec okolo rohovky, viditeľný aj voľným okom, alebo pri očnom vyšetrení. Očná funkcia nebýva poškodená.
Jedinci s Alagillovým syndrómom majú typické črty tváre - hypertelorizmus (hlboko a ďaleko od seba zasadené oči), nízko zasadené uši, prominujúca brada špicatého tvaru, široké čelo.
Medzi ďalšie anomálie patria obličkové anomálie (poškodenie renálnych funkcií, malé obličky), pankreatická nedostatočnosť (zhoršená funkcia), anomálie ciev (môžu spôsobiť ruptúru ciev napr. v mozgu a následnú ruptúru a krvácanie do mozgu.
Diagnostika:
Diagnostika sa stanovuje na základe klinických príznakov, anamnézy pacienta, laboratórnej diagnostiky a zobrazovacích metód, ktoré podrobne určia rozsah poškodenia orgánov. Lekár vysloví podozrenie na Alagillov syndróm v prípade, že sú prítomné 3 z 5 príznakov - poškodenie pečene, kongenitálne chyby srdca, skeletálne abnormality, očné abnormality v kombinácii s prítomnosťou redukcie žlčových odvodných ciest a/alebo typické črty tváre.
Biopsia pečene dokáže redukciu odvodných žlčových kanálikov, čo je prítomná u 90% pacientov.
Genetické vyšetrenie stanoví konečnú diagnózu, ktorá potvrdí prítomnosť mutácie génov.
Liečba:
Liečba je úzko špecifická pre každého pacienta. Nutné je rozpoznať orgánové abnormality, kardiologické defekty, pečeňové poškodenie, obličkové poškodenie.
Kardiológ určí nutnosť chirurgickej liečby. Nutná je dispenzarizácia gastroenterológom, ktorý podáva lieky zlepšujúce odtok žlče do čreva, antihistaminiká vzhľadom na pruritus, hepatoprotektíva, suplementácia nutrientov, vitamínov, riešenie malabsorbcie, podávanie ľahko stráviteľných formúl, pankreatické enzýmy, etc.
- typické črty tváre – široké čelo, prominentná brada u dospelých (prognatia), nízko zasadené uši, oči hlboko a ďaleko od seba (hypertelorizmus), špicatá brada
- kongenitálne srdcové defekty - defekt komorového septa, fallotova tetralógia, stenóza pľúcnych tepien
- poškodenie pečene v rôznom rozsahu (asymptomatické, ľahké až ťažké poškodenie - cirhóza pečene)
- biopsia pečene - nedostatok žlčových kanálov
- hromadenie žlče a kyselín v žlčových cestách (cholestáza)
- hromadenie bilirubínu, cholesterolu v tkanivách
- žltačka, svrbenie kože (pruritus)
- porucha trávania tukov, vitamínov (A,D,E,K), malabsorbcia
- slabé priberanie, slabý rast u novorodencov, neprospievanie
- xantómy (vačky na koži plné cholesterolu)
- posteriorny embryotoxón (zhrubnutý prstenec okolo rohovky)
- obličkové abnormality (malé obličky), poškodenie renálnej funkcie
- pankreatická nedostatočnosť (pankreatopatia)
- vaskulárne abnormality (riziko ruptúry ciev v mozgu a následne krvácanie do mozgu)
- skeletálne abnormality - v oblasti stavcov
- oneskorenie psychomotorického vývoja a kognitívnych funkcií
Pre spojenie sa s ďalšími ľuďmi s rovnakou diagnózou vo vašom okolí sa prihláste.
Prihlásenie